
|
|||
|---|---|---|---|
|
|
Diagnóstico tardio da doença de Wilson: relato de caso Diagnóstico tardío de la enfermedad de Wilson: relato de caso |
|
|
|
*Médico residente de clínica médica do Hospital Universitário Clemente de Faria (HUCF), Montes Claros, MG **Graduanda em Medicina pelas Faculdades Integradas Pitágoras, Montes Claros, MG (Brasil) |
Carolline Santos Aguiar Monção* Bruna Tupinambá Maia** Isis Gabriella Antunes Lopes* Jeniffer Araujo Ribeiro* Thiago Henrique Xavier Guimarães* Adriano Colares Tolentino* |
|
|
|
Resumo A doença de Wilson consiste na diminuição ou ausência da ceruloplasmina, ATPase responsável por intermediar o transporte do cobre no metabolismo das células hepáticas. Desta forma ocorre um acúmulo deste elemento, tendo em vista que a sua excreção na bile encontra-se comprometida. Conseqüentemente há morte hepatocitária e liberação de grande quantidade de cobre no sangue periférico, o qual provoca hemólise e impregnação dos núcleos da base no sistema nervoso, córnea, fígado e rins. Sendo assim, o quadro clínico desta irá abranger manifestações neurológicas, psiquiátricas, hepáticas e oculares. O presente artigo relata o caso de um paciente portador desta enfermidade, cujo diagnóstico foi feito tardiamente. Unitermos: Doença de Wilson. Diagnostico tardio. Relato de caso.
Recepção: 14/08/2014 - Aceitação: 29/11/2014.
|
|||
|
|
EFDeportes.com, Revista Digital. Buenos Aires, Año 19, Nº 199, Diciembre de 2014. http://www.efdeportes.com/ |
|
|
1 / 1
Introdução
A Doença de Wilson (DW) é uma entidade conhecida há mais de um século, sendo inicialmente denominada de pseudoesclerose por Westphal em 1883. Em 1898, Strumpell, evidenciou acometimento hepático em autópsia de dois paciente portadores de tremores. Mas somente em 1912, Kinnier Wilson descreveu a doença e suas alterações hepáticas e neurológicas, chamando-a inicialmente de degeneração lenticular progressiva. Hall, em 1921, a descreveu como degeneração hepatolenticular.1,2,3
A doença de Wilson, como hoje é chamada, é incomum, com inicio entre 11 e 25 anos de idade, sendo transmitida por herança autossômica recessiva. Com a herança, o paciente não forma ou forma com deficiência uma ATPase transportadora de cobre, a ceruloplasmina, levando a dificuldade de eliminar o cobre na bile. Isso resulta na saturação do armazenamento hepático de cobre com posterior morte hepatocitária e liberação de grande quantidade de cobre no sangue periférico, culminando em hemólise e impregnação dos núcleos da base do cérebro, córnea, fígado e rins. Sendo assim, o quadro clínico da DW irá abranger manifestações neurológicas, psiquiátricas, hepáticas e oculares. Para o diagnóstico da DW, devemos direcionar a atenção para o anel de Kayser-Fleischer, as alterações nos níveis séricos de ceruloplasmina e ao exame de excreção urinária de cobre. Importante destacar o papel da Ressonância Nuclear Magnética (RNM) cerebral na DW, que pode demonstrar a presença de alguns sinais característicos da doença, como as “Faces de Panda”, que contribuem para a elucidação diagnóstica.4,5,6,7
Relato do caso
Paciente J.L.S.M., sexo masculino, 21 anos, solteiro, balconista, leucoderma, apresentou-se para consulta médica com quadro arrastado – aproximadamente 2 anos – de fraqueza generalizada, associado à dificuldade de deambulação, desânimo, tristeza, exclusão social,dificuldade para falar e engasgos esporádicos ao alimentar-se. Sem epigastralgia, febre ou perda de peso.
Paciente sem comorbidades prévias, negou tabagismo, cirurgias, uso de medicação contínua, contato com águas naturais ou internações prévias. Etilista social. Durante a infância, desenvolveu-se nos padrões normais, apesar de pouco introspectivo.
História familiar sem informações importantes.
Moradia com boa infra-estrutura em zona urbana.
Dados vitais e exame físico normal, exceto presença de rigidez muscular, bradicinesia e instabilidade postural.
Foi iniciada então investigação para doenças neuromusculares que levam a fraqueza e rigidez muscular. Primeiro foram averiguados os exames que o paciente trazia de casa, realizado em consultas médicas anteriores, dentre eles, hemogramas, função hepática, função renal, eletrólitos, EAS, culturas de urina e sangue, exames de fezes, provas de atividade inflamatória, CPK, sorologias para Hepatites virais e HIV, FAN, eletroforese de proteínas, anti-receptores de acetilcolina, exames de imagem como ultrassom de abdome superior, EDA e Ecocardiograma todos normais.
Paciente recebeu diagnóstico prévio de miastenia gravis, sendo realizado tratamento com neostigmina por 01 ano, sem sucesso. Foi quando irmão do paciente procurou por outros profissionais, questionando a não melhora do quadro clínico do paciente.
Foi solicitado durante o acompanhamento atual dosagem de Ceruloplasmina sérica, Cobre sérico, função hepática, novo ultrassom de abdome superior, cobre urinário, exame oftalmológico e RNM de encéfalo. Ao exame oftalmológico houve relato de quadro de depósito corneano sugestivo de cobre (Anel de Kayser-Fleischer). Ceruloplasmina de 2,9 mg/dl ;Cobre sérico de 22 ng/dl; Cobre urinário de 29,1 ng/24h. Ultrassom de abdome: fígado de contornos lobulados e parenquima difusamente heterogêneo, com padrão micronodular difuso, compatível com a presença de hepatopatia crônica fibrosante; espessamento e aumento da ecogenicidade periportal; esplenomegalia leve. Eletroneuromiografia: Normalidade dos achados eletroneuromiográficos. A ressonância magnética: hiperintensidades nas seqüências ponderadas em T2 ao nível do mesencéfalo, núcleos lentiformes e porções posteriores da cápsula interna; Hiperintensidade nas seqüências Flair e T2 ao nível do esplênio do corpo caloso (figura 01).
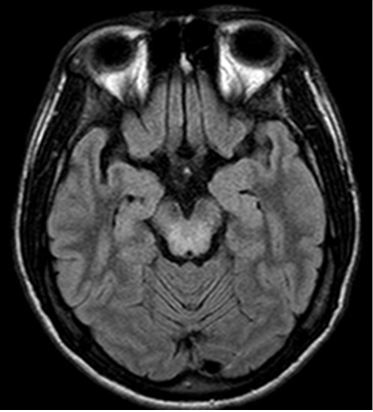
Figura 01. Sinal da “faces de panda”
Ao associar os achados clínico-laboratoriais, com os resultados do exame oftalmológico, eletroneuromiografia e RNM, concluímos o diagnóstico de Doença de Wilson e iniciado o tratamento com D-penicilamina associado a medidas higienodietéticas. O paciente evolui bem, com regressão lenta e progressiva dos sintomas neuro-psiquiátricos e o controle laboratorial seriado evidenciou incremento da cuprúria.
Discussão
Fica claro perceber com este caso a gama de diagnósticos diferenciais que podem surgir quando o paciente abre o quadro de DW com manifestações predominantemente neurológicas, sem alterações nos exames de função hepática ou renal, nos levando à investigação de doenças neuromusculares que, por vezes, podem atrasar o diagnóstico. Além disso, percebemos que a identificação do anel de Kayser-Fleischer pode não ser percebido em uma primeira avaliação. Ao levantarmos a hipótese de uma doença neuromuscular, com eletroneuromiografia dentro dos limites da normalidades, e associarmos com o padrão clínico de rigidez muscular, bradicinesia e instabilidade postural, além de imagem hepática compatível com acometimento parenquimatoso difuso, passamos a acreditar fortemente em DW. A hipótese foi confirmada ao associarmos todos esses achados citados com o resultado da RNM e as dosagens de cobre sérico e ceruloplasmina.
A DW, apesar de classicamente ser composta por uma constelação de manifestações neurológico-psiquiátricas, hepáticas, renais e corneanos, pode, em certas ocasiões, apresentar-se com quadro clínico mais direcionado para um ou outro órgão (neste caso, manifestações predominantemente neurológicas e corneanas), sendo o exame sistêmico e freqüente do paciente de suma importância para flagrarmos as manifestações da deposição de cobre em outros órgãos. Além da necessidade de uma avaliação oftalmológica cuidadosa, por vezes, com auxilio do oftalmologista, precisamos estar também familiarizados com as alterações típicas da DW na RNM.
Referências
-
McDowell FH, Lee JE, Sweet RD. Extrapiramidal disease. In Baker AB (ed). Clinical neurology. Philadelphia: Harper & Row, 1978:38, 1-67.
-
Le Coz P, Goldstein B, Woimant F, Haguenau M. Maladie de Wilson. Editions Techniques. Encyl Méd Chir. (Paris-France), Neurologie, 17060 A10, 1992,1-10.
-
Brito JCF, Coutinho MAP, Almeida HJF, Nóbrega PV. Doença de Wilson: diagnóstico clínico e sinais da das “Faces do Panda” à ressonância magnética – relato de caso. Arq Neuropsiquiatr, 2005; 63(1): 176-179
-
Silva AC, Colósimo AP, Salvestro D. Doença de Wilson (degeneração hepatolenticular): revisão bibliográfica e relato de caso. Rev Méd Minas Gerais. 2010; 20(N. Esp.): 404-411.
-
Korman JD, Volenberg I, Balko J, Webster J, Schiodt FV, Squires RH Jr, Fontana RJ, Lee WM, Schilsky ML. Pediatric and Adult Acute Liver Failure Study Groups. Hepatology. 2008 Oct; 48(4):1167-74.
-
Seo JK. Pediatr Gastroenterol Hepatol Nutr. 2012 Dec; 15(4):197-209. Epub 2012 Dec 31.
-
Rosencrantz R, Schilsky M. Semin Liver Dis. 2011 Aug; 31(3):245-59. Epub 2011 Sep 7.
Outros artigos em Portugués
| |

Búsqueda personalizada
|
|---|---|
|
EFDeportes.com, Revista
Digital · Año 19 · N° 199 | Buenos Aires,
Diciembre de 2014 |
|