Introdução
A prevalência da obesidade vem apresentando um aumento em vários países ao
redor do mundo nas últimas décadas. Esta constatação é importante já que o
excesso de gordura corporal, sobretudo a abdominal, está diretamente
relacionado com alterações do perfil lipídico, com o aumento da pressão
arterial e a hiperinsulinemia, considerados fatores de risco para o
desenvolvimento de doenças crônicas, como o diabetes melito tipo 2
(DM2) e as doenças cardiovasculares (Ciolac, Guimarães, 1997). Níveis
elevados de leptina e de ácido úrico e a alteração dos fatores
fibrinolíticos também têm sido observados em indivíduos obesos (Chejab e
colaboradores, 1996), aumentando os fatores de descontrole de peso. O conjunto
destas alterações tem sido descrito como síndrome metabólica
(Oliveira e colaboradores, 2004).
No senso comum, até pouco tempo atrás, acreditava-se que a DM2 era uma doença
manifestada por circunstância isolada. Como dito, hoje já se sabe que há um
conjunto de fatores e patologias que podem estar envolvidos na manifestação da
DM2. A síndrome metabólica – também conhecida como síndrome X, síndrome
da resistência à insulina, quarteto mortal ou síndrome plurimetabólica
(Figura 1) – é caracterizada pelo agrupamento de fatores de risco
cardiovascular como hipertensão arterial, resistência à insulina,
hiperinsulinemia, intolerância à glicose/DM2, obesidade central e dislipidemia
(LDL-colesterol alto, triglicérides alto e HDL-colesterol baixo) (Ciolac,
Guimarães, 1997).
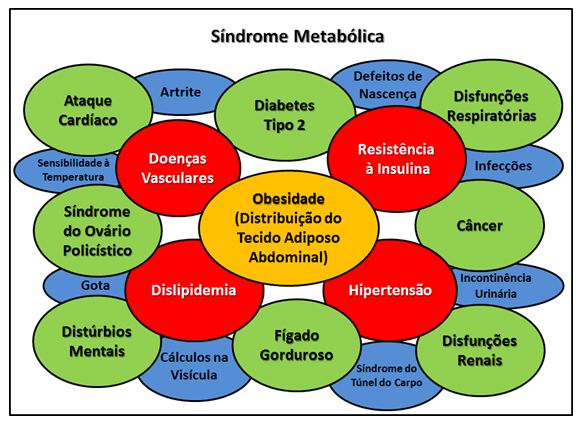
Figura
1. Doenças associadas à Síndrome Metabólica e suas complicações
secundárias
Estudos epidemiológicos e clínicos têm demonstrado que a prática regular de
atividade física é um importante fator para a prevenção e tratamento desta
doença (Duncan e colaboradores, 1992). A obesidade, até então vista como uma
desordem primariamente de alta ingestão energética, hoje, levando-se em conta
os princípios questionados pelos estudos sobre qualidade de vida, tende também
a ser tratada como uma doença prevalente pelo baixo gasto energético que ao
alto consumo calórico, surgindo a inatividade física como o maior fator
etiológico para o crescimento desta doença nos grandes centros urbanos
(Erikson e colaboradores, 1997). O objetivo principal desta revisão é a
abordagem dos mecanismos de ação da instalação da resistência à insulina e
os efeitos positivos que o exercício físico podem desencadear no tecido
musculoesquelético.
Materiais
e métodos
A fundamentação teórica foi conduzida por uma pesquisa do tipo
bibliográfica, pois esta permitiu a oportunidade de adquirir amplas
informações relativas aos mecanismos de instalação da síndrome metabólica
(resistência à insulina) e as adaptações induzidas pelo exercício físico.
De acordo com Marconi e Lakatos (1988, p. 57-58), a pesquisa bibliográfica tem
como finalidade “colocar o pesquisador em contato direto com tudo aquilo
que foi escrito sobre determinado assunto”. Apesar de este estudo envolver
o conhecimento biológico, o qual tem como base e tradição sua demonstração
por métodos de investigação laboratorial, teve-se a necessidade, em relação
à temática escolhida, de se construir um referencial alicerçado em teorias e
pressupostos experimentais disponíveis na literatura, para que houvesse uma
interpretação sob um “olhar” docente, preocupado em possibilitar ao
graduando um contato numa perspectiva pedagógica.
Mecanismos
de instalação da resistência à insulina
A insulina é uma proteína com duas cadeias polipeptídicas ligadas, contendo
21 aminoácidos na cadeia A e 30 aminoácidos na cadeia B. Ela é um hormônio e
um neurotransmissor. São vários os tipos de estímulos e os fatores de
inibição da liberação da insulina pelas células β do pâncreas,
localizadas nas ilhotas de Largerhans. O principal estímulo é a taxa de
glicose no sangue e a presença de alimentos no estômago estimula a produção
de enzimas que levam à secreção de insulina.
A insulina sérica se liga a um receptor específico na superfície de suas
células-alvo. O receptor é um grande complexo glicoprotéico transmembrana que
pertence à superfamília de receptores tipo 3 ligados a quinases e constituindo
em duas subunidades alfa e duas beta. Os receptores ocupados se agregam em
grupos, que são interiorizados em vesículas, resultando em infra-regulação.
A insulina interiorizada é degradada nos lisossomos, mas os receptores são
reciclados para a membrana plasmática (Kahn, 1985; Kasuga e colaboradores,
1982). O ATP age como doador de fosfatos e a fosforilação ocorre em resíduos
tirosina. O mecanismo molecular exato da ação da insulina é desconhecido, mas
parece depender da remoção do efeito inibitório da subunidade α sobre a
atividade da subunidade β do seu receptor (Figura 2).
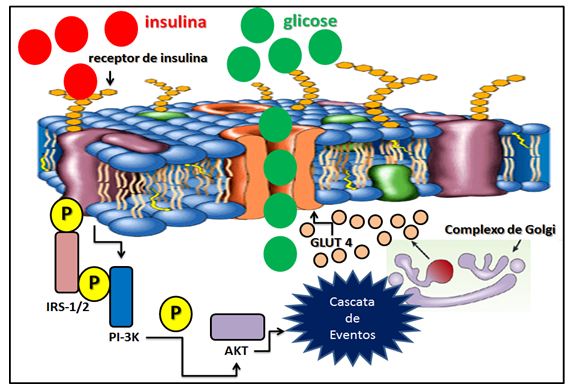
Figura
2. Via de sinalização da insulina na captação da glicose
O receptor de insulina possui dois substratos (IRS-1/2) que sofrem rápida
fosforilação nos grupos tirosina em reposta à insulina (15). A fosforilação
da tirosina permite sua associação a proteínas que possuem domínios
sulfidrilas (SH) 2 e 3 de reconhecimento específico para fosfotirosina. As
proteínas IRS-1/2 desempenham função essencial na transmissão do sinal
insulínico e a fosforilação desses substratos permite a interação com
diversas proteínas adaptadoras ou com atividade enzimática, caracterizando o
efeito pleiotrópico da insulina, desencadeando uma cascata de eventos
pró-ativação GLT-4. Observa-se grande associação entre a enzima
fosfatidilinositol 3-quinase (PI-3K) com IRS-1/2 após estimulação com
insulina (Folli e colaboradores, 1992). A PI-3K contém dois sítios SH2 e um
sítio SH3, apresentando papel importante em diversos processos celulares,
incluindo a regulação da mitogênese, diferenciação celular e transporte de
glicose estimulada pela insulina. A ativação da PI-3K aumenta a fosforilação
em serina da proteína quinase B (AKT), permitindo o transporte de glicose no
músculo e no tecido adiposo por meio da translocação do GLUT4 para a membrana
celular. Desta forma, a ativação da AKT resulta na translocação do GLUT4
para a membrana, permitindo a entrada da glicose por difusão facilitada (Backer
e colaboradores, 1992; Boschero, 1996).
A resistência à ação da insulina no músculo esquelético é uma das
principais características em indivíduos diabéticos. Embora o número de
pesquisas nessa área tenha crescido exponencialmente, os mecanismos
responsáveis pela instalação dessa patologia ainda não foram esclarecidos.
Porém, há um consenso da existência de uma forte correlação entre
resistência à insulina, inatividade e elevado conteúdo de lipídios
intracelulares (Jenkins e colaboradores, 1988; Hawley e colaboradores, 2000;
Hansson e colaboradores, 2005; Savage e colaboradores, 2005; Silveira e
colaboradores, 2008). Experimentalmente, esse quadro tem sido facilmente
induzido em animais submetidos à dieta hiperlipídica (Brehn e colaboradores,
2006; Chanseaume e colaboradores, 2006; Choi e colaboradores, 2007; Silveira e
colaboradores, 2007) ou células isoladas expostas a altas concentrações de
nutrientes incluindo ácidos graxos (Hirabara e colaboradores, 2007; Sabin e
colaboradores, 2007; Lambertucci e colaboradores, 2008).
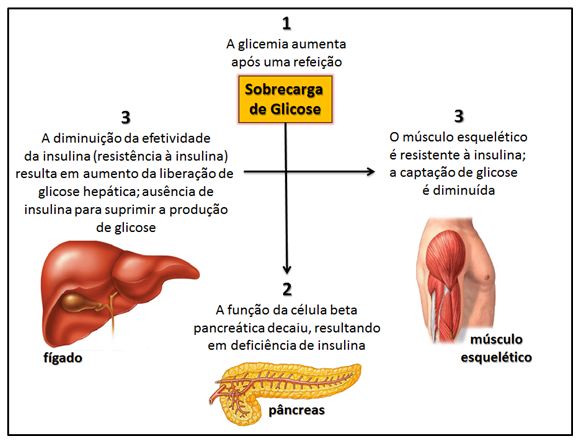
Figura
3. Eventos fisiopatológicos que levam à hiperglicemia em pacientes com
diabetes do tipo 2
Randle e colaboradores (1963) foram os primeiros a demonstrarem que, sob elevada
exposição aos ácidos graxos, a utilização de glicose é substancialmente
reduzida nos tecidos periféricos incluindo o muscular (Figura 3). Este
mecanismo foi originalmente proposto como ciclo glicose-ácido graxo. Na última
década, um número crescente de estudos mostrou ainda a existência de
mecanismos moleculares associados à resistência à insulina. O DM2 induz
inibição de componentes da via de sinalização da insulina causando
prejuízos na transmissão do sinal deste hormônio (Jenkins e colaboradores,
1988; Hawley e colaboradores, 2000; Silveira e colaboradores, 2008; Chanseaume e
colaboradores, 2006; Silveira e colaboradores, 2007; Hirabara e colaboradores,
2007), sugerindo, portanto, que, a resistência à insulina pode estar associada
a uma menor resposta durante o processo de fosforilação e ativação dos
transportadores de glicose pela insulina. Observações recentes demonstraram
que este mecanismo está acompanhado de uma baixa atividade mitocondrial e de
aumento na produção de espécies reativas de oxigênio (EROs), induzindo
estresse oxidativo (Savage e colaboradores, 2007; Griffin e colaboradores, 1999;
Rezende e colaboradores, 2004) (Figura 4).
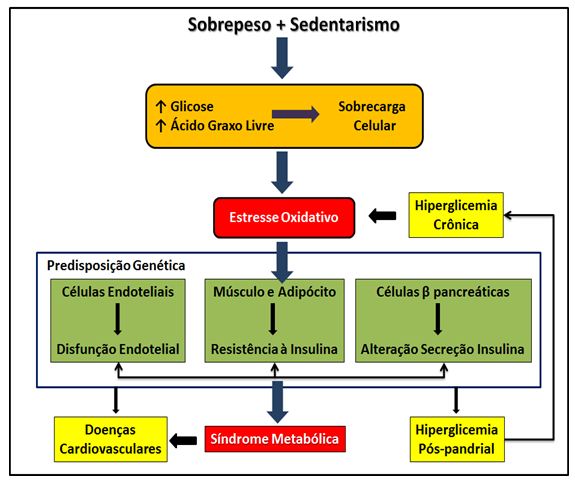
Figura
4. Fatores metabólicos envolvidos no aumento do estresse oxidativo e
desencadeamento da síndrome metabólica
Estresse oxidativo pode ser definido como o desequilíbrio causado pelo excesso
na produção das EROs associado à queda na capacidade antioxidante celular
(Sies, 1997; Finkel, 2003; Scandalios, 2005). As EROs podem ser derivadas de
fontes exógenas ou produzidas endogenamente como conseqüência normal das
funções celulares, por diversas enzimas que utilizam o oxigênio (O2) como
substrato (Finkel, Holbrook, 2000; Finkel, 2003). Resultam da excitação do O2
para formar oxigênio singleto (1O2) ou da redução do O2, nas mitocôndrias,
pela citocromo oxidase (aa3) – que catalisa a transferência de um, dois e
três elétrons – originando, respectivamente, radical superóxido (O2-),
peróxido de hidrogênio (H2O2) e radical hidroxila (OH∙). Este processo
de redução produz água (H2O) como produto final da reação (Gutteridge,
1995; Sies, 1997). As principais EROs dividem-se em radicalares e
não-radicalares. Podem, ainda, reagir com outros átomos e formar espécies
reativas, como o peroxinitrito (OONO-) – formado a partir da reação do
óxido nítrico (NO) com O2- (Giasson e colaboradores, 2002). Algumas destas
espécies são extremamente instáveis, enquanto outras são livremente difusas
e de meia-vida relativamente longa (Gutteridge, 1995). Por serem extremamente
reativas, níveis elevados ou sustentados de EROs podem causar danos severos ao
DNA (deleções, mutações e translocações; além de oxidação de
filamentos), às proteínas (modificações locais específicas nos
aminoácidos, fragmentação da corrente do peptídeo e aumento da
susceptibilidade da proteólise) e aos lipídios (alterações em propriedades
de membranas biológicas) (Giasson e colaboradores, 2002; Scandalios, 2005).
Tais circunstâncias podem conduzir à necrose ou apoptose, além de
sinalização para ativação gênica de eventos patológicos que até então
estavam “dormentes”. A literatura aponta que diversas doenças
neuromusculares, neurodegenerativas e até mesmo o diabetes potencializam a
ação celular de gerar as EROs (Butler e colaboradores, 2003).
Curiosamente, indivíduos treinados fisicamente exibem um elevado conteúdo
intramuscular de ácidos graxos (Van Loon, Goodpaster, 2006). Porém, ao
contrário de indivíduos sedentários, apresentam elevada capacidade
mitocondrial e antioxidante, sendo, portanto, altamente sensíveis à insulina e
sugerindo que a baixa capacidade mitocondrial associada ao aumento de espécies
reativas de oxigênio são mesmo dois importantes fatores indutores de
resistência à insulina (Savage e colaboradores, 2007; Silveira e
colaboradores, 2008; Griffin e colaboradores, 1999; Rezende e colaboradores,
2004).
Exercício
físico e adaptações moleculares dos transportadores de glicose
O transporte de glicose através da membrana celular é um fator limitante na
utilização desta molécula pelo músculo durante o exercício físico (Cline e
colaboradores, 1999). O exercício físico e a insulina são os agentes
fisiológicos mais importantes no transporte de glicose no músculo
esquelético. Estudos indicam que o exercício físico agudo não tem capacidade
de elevar a fosforilação em tirosina do receptor de insulina e nem de aumentar
a fosforilação em tirosina do IRS-1 estimulada por insulina (Henriksen, 2002);
porém, configura-se que o exercício potencializa o efeito da insulina na
fosforilação do IRS-2 com concomitante aumento da PI-3K (Howlett e
colaboradores, 2002). Wojtaszewski e colaboradores (1999) pontuam ainda que o
exercício induz maior fosforilação em serina da AKT – proteína fundamental
para o início da translocação do GLUT4 para a membrana citoplasmática.
No exercício físico, a contração muscular aumenta a translocação de GLUT4
independente da presença de insulina. Sugere-se que isso ocorra em virtude do
aumento da concentração intracelular de cálcio ([Ca2+]i). A liberação de
cálcio das cisternas do retículo sarcoplasmático, mediada pela
despolarização (necessária para a formação do complexo actina-miosina), é
observada como uma hipótese para a mediação do transporte da glicose. Tal
sugestão relaciona-se na observação de que o aumento do transporte de glicose
correlaciona-se com a freqüência da contração e não com a duração ou
tensão do movimento (Etgen e colaboradores, 1993). O mecanismo proposto seria
de que o cálcio no citoplasma poderia facilitar ou desencadear a ativação de
estruturas moleculares envolvidas na cascata de sinalização intracelulares, as
quais promovem os efeitos imediatos e prolongados do exercício sobre o
transporte de glicose. Uma molécula bem estudada é a proteína quinase C
(cálcio-dependente e sinalizadora intermediária), que é ativada pela
contração muscular e parece estar envolvida na regulação do transporte de
glicose estimulado pela contração muscular (Gomes e colaboradores, 2005).
O principal mecanismo pelo qual insulina e exercício físico efetuam o
transporte de glicose no músculo esquelético se dá por meio da translocação
do GLUT4, por ser o maior transportador de glicose e expresso no próprio
músculo (Hayashi e colaboradores, 1997; Goodyear, Kahn, 1998). A contração
muscular estimula a translocação do GLUT4 até mesmo na ausência ou
insuficiência de insulina (Hayashi e colaboradores, 1997; Goodyear, Kahn,
1998); porém, estudos indicam que há diferentes núcleos intracelulares de
GLUT4 (um estimulado por insulina e outro pelo exercício físico), supondo que
a insulina e o exercício físico ativam GLUT4 por diferentes mecanismos (Douen
e colaboradores, 1990; Coderre e colaboradores, 1995).
A sensibilidade à insulina pode ser elevada com a prática sistemática do
exercício físico, independente da condição de composição corporal do
indivíduo, sugerindo que o principal efeito do exercício seja o aumento da
expressão de estruturas intracelulares da via de sinalização da insulina,
principalmente dos transportadores de glicose na célula músculo esquelética
(O'Donovan e colaboradores, 2005; Teran-Garcia e colaboradores, 2005). Também
se relata que o treinamento físico diminui a adiposidade, o tamanho da célula
de gordura, os níveis de insulina no plasma e aumenta a expressão de GLUT4 no
músculo, favorecendo, assim, um estímulo importante para o transporte de
glicose estimulada por insulina (Hardman, 1996; Mensink e colaboradores, 2003;
Stubbs, Lee, 2004).
O aumento de sensibilidade à insulina em sujeitos que apresentam resistência a
tal hormônio pode ocorrer por intermédio da expressão gênica e do conteúdo
de GLUT4, bem como a ativação das proteínas intracelulares envolvidas nas
vias sinalizadoras de translocação das vesículas de GLUT4. Exercícios
físicos praticados de forma aguda podem promover tanto o aumento da expressão
gênica quanto do conteúdo de GLUT4, o que resulta no aumento de translocação
dos receptores de glicose nas células musculares. Exercícios de natação,
comparados a corrida em esteira, foram capazes de promover manutenção da
diminuição da resistência à insulina por mais tempo. Já exercícios de alta
intensidade e curta duração são relatados como mais eficientes do que
atividades de baixa intensidade e longa duração (Ribeiro e colaboradores,
2011).
Concluindo, há fortes evidências de que a prática de exercícios físicos
pode reverter os efeitos negativos promovidos pela resistência à insulina. O
mais evidente são os estudos que indicam que a prevenção contra a obesidade e
a prática de exercícios inserida na rotina das pessoas são os melhores meios
para prevenção da instalação das patologias envolvidas na síndrome
metabólica.
Referências
bibliográficas
-
Ciolac,
E. G.; Guimarães, G. V. Exercício físico e síndrome metabólica. Rev.
Bras. Med. Esporte, v. 10, p. 319-324, 2004.
-
Chehab,
F. F.; Lim, M. E.; Lu, R. Correction of the sterility defect in homozygous
obese females mice by treatment with the human recombinant leptin. Nat.
Genet., v. 12, p. 318-320, 1996.
-
Oliveira,
C. L.; Mello, M. T.; Cintra, I. P.; Fisberg, M. Obesidade e síndrome
metabólica na infância e adolescência. Rev. Nutr., v. 17, n. 2, p.
237-245, 2004.
-
Duncan,
B. B.; Schmidt, M. I.; Polanczyk, C. A.; Menegue, S. Altos coeficientes de
mortalidade em populações adultas brasileiras: uma comparação
internacional. Rev. Assoc. Med. Bras., v. 38, p. 138-144, 1992.
-
Eriksson,
J.; Taimela, S.; Koivisto, V. A. Exercise and the metabolic syndrome.
Diabetologia, v. 40, p. 124-135, 1997.
-
Marconi,
M. A.; Lakatos, E. M. Técnicas de Pesquisa. São Paulo: Atlas, 1986.
-
Kahn
CR. Current concepts of the molecular mechanism of insulin action. Ann.
Rev. Med., v. 36, p. 429-451, 1985.
-
Kasuga,
M.; Karlsson, F. A.; Kahn, C. R. Insulin stimulates the phosphorylation of
the 95,000-dalton subunit of its own receptor. Science, v. 215, p.
185-187, 1982. 1982.
-
Folli
F.; Saad, M. J. A.; Backer, J. M.; Kahn, C. R. Insulin stimulation of
phosphatidylinositol 3-kinase and association with insulin receptor
substrate 1 in liver and muscle of the intact rat. J. Biol. Chem., v.
267, p. 22171-22177, 1992.
-
Backer,
J. M.; Myers, M. G. Jr; Shoelson, S. E.; Chin, D. J.; Sun, X. J.; Miralpeix,
M. et al. Phosphatidylinositol 3’-kinase is activated by association with
IRS-1 during insulin stimulation. EMBO J., v. 11, p. 3469-3479, 1992.
-
Czech,
M. P.; Corvera, S. Signaling mechanisms that regulate glucose transport.
J. Biol. Chem., v. 274, p. 1865-1868, 1999.
-
Matschinsky,
F. M. Banting Lecture 1995: a lesson in metabolic regulation inspired by the
glucokinase glucose sensor paradigm. Diabetes, v. 45, p. 223-241,
1996.
-
Boschero,
A. C. Acoplamento excitação-secreção nas células B pancreáticas. Arq.
Bras. Endocrinol. Metab., v. 40, p. 149-155, 1996.
-
Jenkins,
A. B.; Storlien, L. H.; Chisholm, D. J.; Kraegen, E. W. Effects of nonesterified
fatty acid availability on tissue-specific glucose utilization in rats
in vivo. J. Clin. Invest., v. 82, p. 293-299, 1988.
-
Hawley,
J. A.; Burke, L. M.; Angus, D. J.; Fallon, K. E.; Martin, D. T. et al.
Effect of altering substrate availability on metabolism and performance
during intense exercise. Br. J. Nutr., v. 84, p. 828-838, 2000.
-
Hansson,
O.; Donsmark, M.; Ling, C.; Nevsten, P.; Danfelter, M. et al. Transcriptome
and proteome analysis of soleus muscle of hormone-sensitive lipase-null
mice. J. Lipid. Res., v. 46, p. 2614-2623, 2005.
-
Savage,
D. B.; Petersen, K. F.; Shulman, G. L. Disordered lipid metabolism and the
pathogenesis of insulin resistance. Physiol. Rev., v. 87, p. 507-520,
2007.
-
Silveira,
L. R.; Fiamoncini, J.; Hirabara, S. M.; Procópio, J.; Cambiaghi, T. D. et
al. Updating the effects of fatty acids on skeletal muscle. J. Cell
Physiol., v. 217, p. 1-12, 2008.
-
Brehm,
A.; Krssak. M.; Schmid, A. I.; Nowotny, P.; Waldhausl, W. et al. Increased
lipid availability impairs insulin-stimulated ATP synthesis in human
skeletal muscle. Diabetes, v. 55, p. 136-140, 2006.
-
Chanseaume,
E.; Bielicki, G.; Tardy, A. L.; Renou, J. P.; Freyssenet, D. et al. Impaired
resting muscle energetics studied by 31P-NMR in diet-induced obese
rats. J. Nutr., v. 2194-2200, 2006.
-
Choi,
C. S.; Savage, D. B.; Abu-Elheiga, L.; Liu, Z. X.; Kim, S. et al. Continuous
fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total
energy expenditure, reduces fat mass, and improves insulin sensitivity.
Proc. Natl. Acad. Sci. U.S.A., v. 104, p. 16480-16485, 2007.
-
Silveira,
L. R.; Hirabara, S. M.; Alberici, L. C.; Lambertucci, R. H.; Peres, C. M. et
al. Effect of Lipid Infusion on Metabolism and Force of rat skeletal muscles
during intense contractions. Cell. Physiol. Biochem., v. 20, p.
213-226, 2007.
-
Hirabara,
S. M.; Silveira, L. R.; Abdulkader, F.; Carvalho, C. R. O.; Procópio, J.
Time-dependent effects of fatty acids on skeletal muscle metabolism. J.
Cell Physiol., v. 210, p. 7-15, 2007.
-
Sabin,
M. A.; Stewart, C. E. H.; Crowne, E. C.; Turner, S. J.; Hunt, L. P. et al.
Fatty acid-induced defects in insulin signalling, in myotubes derived from
children, are related to ceramide production from palmitate rather than the
accumulation of intramyocellular lipid. J. Cell Physiol., v. 211, p.
244-252, 2007.
-
Lambertucci,
R. H.; Hirabara, S. M.; Silveira, L. R.; Levada-Pires, A. C.; Curi, R. et
al. Palmitate increases superoxide production through mitochondrial electron
transport chain and NADPH oxidase activity in skeletal muscle cells. J.
Cell Physiol., v. 216, p. 796–804, 2008.
-
Randle,
P. J.; Garland, P. B.; Hales, C. N.; Newsholme, E. A. The glucose fatty-acid
cycle. Its role in insulin sensitivity and the metabolic disturbances of
diabetes mellitus. Lancet I., v. 13, p. 785-789, 1963.
-
Griffin,
M. E.; Marcucci, M. J.; Cline, G. W. Free fatty acid-induced insulin
resistance is associated with activity of protein kinase C theta and
alterations in the insulin signaling pathway. Diabetes, v. 48, p.
1270-1274, 1999.
-
Rezende,
E. A.; Sampaio, I. B. M.; Ishitani, L. H. Causas múltiplas de morte por
doenças crônico-degenerativas: uma análise multidimensional. Cad.
Saúde Pública, v. 20, p. 1223-1231, 2004.
-
Sies,
H. Oxidative stress: oxidants and antioxidants. Exp. Physiol., v. 82,
p. 291-295, 1997.
-
Finkel,
T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol., v.
15, p. 247-254, 2003.
-
Scandalios,
J. G. Oxidative stress: molecular perception and transduction of signals
triggering antioxidant gene defenses. Braz. J. Med. Biol. Res., v.
38, p. 995-1014, 2005.
-
Finkel,
T.; Holbrook, N. J. Oxidants, oxidative stress and the biology of ageing.
Nature, v. 408, p. 239-247, 2000.
-
Gutteridge,
J. M. C. Lipid peroxidation and antioxidants as biomarkers of tissue damage.
Clin. Chem., v. 41, p. 1819-1828, 1995.
-
Giasson,
B. I.; Ischiropoulos, H.; Lee, V. M.; Trojanowski, J. Q. The relationship
between oxidative/nitrative stress and pathological inclusions in Alzheimer’s
and Parkinson’s Diseases. Free Rad. Biol. Med., v. 32, p.
1264-1275, 2002.
-
Butler
AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell
Deficit and increased β-Cell apoptosis in humans with type 2 diabetes. Diabetes,
v. 52, p. 102-110, 2003.
-
Van
Loon, L. J.; Goodpaster, B. H. Increased intramuscular lipid storage in
the insulin-resistant and endurance-trained state. Pflügers Arch.,
v. 451, p. 606-616, 2006.
-
Cline,
G. W.; Petersen, K. F.; Krssak, M.; Shen, J.; Hundal, R. S. et al. Impaired
glucose transport as a cause of decreased insulinstimulated muscle glycogen
synthesis in type 2 diabetes. New Engl. J. Med., v.341, p. 240-246,
1999.
-
Henriksen,
E. J. Invited review: Effects of acute exercise and exercise training on
insulin resistance. J. Appl. Physiol., v.93, n.2, p.788-796, 2002.
-
Wojtaszewski,
J. F.; Higaky, Y.; Hirshman, M. F.; Michael, M. D.; Dufresne, S. D. et al.
Exercise modulates postreceptor insulin signaling and glucose transport in
muscle-specific insulin receptor knockout mice. J. Clin. Invest.,
v.104, p.1257-1264, 1999.
-
Etgen,
G. J. Jr.; Brozinick, J. T. Jr.; Kang, H. Y.; Ivy, J. L. Effects of exercise
training on skeletal muscle glucose uptake and transport. Am. J. Physiol.,
v. 264, p. 723-733, 1993.
-
Gomes,
M. R.; Rogero, M. M.; Tirapegui, J. Considerações sobre cromo, insulina e
exercício físico. Rev. Bras. Med. Esporte, v. 11, p. 262-266, 2011.
-
Hayashi,
T.; Wojtaszewski, J. F.; Goodyear, L. J. Exercise regulation of glucose
transport in skeletal muscle. Am. J. Physiol., v.273, p.E1039-1051,
1997.
-
Goodyear,
L. J.; Kahn, B. B. Exercise, glucose transport, and insulin sensitivity. Ann.
Rev. Med., v.49, p.235-261, 1998.
-
O'Donovan,
G.; Kearney, E. M.; Nevill, A. M.; Woolf-May, K.; Bird, S. R. The effects of
24 weeks of moderate- or high-intensity exercise on insulin resistance.
Eur. J. Appl. Physiol., v., p.1-7, 2005.
-
Coderre,
L.; Kandror, K. V.; Vallega, G.; Pilch, P. F. Identification and
characterization of an exercise sensitive pool of glucose transporters in
skeletal muscle. J. Biol. Chem., v.270, p.27584- 27588, 1995.
-
Douen,
A. G.; Ramlat, T.; Rastogi, S.; Bilan, P. J.; Cartee, G. D. et al. Exercise
induces recruitment of the "insulin-responsive glucose
transporter". Evidence for distinct intracellular insulin- and
exercise-recruitable transporter pools in skeletal muscle. J. Biol. Chem.,
v.265, p.13427-13430, 1990.
-
Teran-Garcia,
M.; Rankinen, T.; Koza, R. A.; Rao, D. C.; Bouchard, C. Endurance
training-induced changes in insulin sensitivity and gene expression. Am.
J. Phys. Endocrinol. Metab., v.288, p.E1168-78, 2005.
-
Hardman,
A. E. Exercise in the prevention of atherosclerotic, metabolic and
hypertensive diseases: a review. J. Sports Sci., v.14, p.201-218,
1996.
-
Mensink,
M.; Blaak, E. E.; Vidal, H.; De Bruin, T. W.; Glatz, J. F. et al. Lifestyle
changes and lipid metabolism gene expression and protein content in skeletal
muscle of subjects with impaired glucose tolerance. Diabetologia,
v.46, p.1082-1089, 2003.
-
Stubbs,
C. O.; Lee, A. J. The obesity epidemic: both energy intake and physical
activity contribute. Med. J. Australia, v.181, p.489-491, 2004.
-
Ribeiro,
H. Q. T,; Camargo, R. G.; Lima, W. P.; Zanuto, R.; Carnevali Júnior, L. C.
Adaptações agudas promovidas por exercícios no aumento da expressão
gênica, conteúdo e translocação da proteína GLUT-4 no músculo
esquelético e melhora na responsividade à insulina. Rev. Bras. Fisiol.
Exerc., v. 10, p. 106-110, 2011.
Outros artigos em
Portugués
 |
|
|
EFDeportes.com, Revista
Digital · Año 17 · N° 172 | Buenos Aires,
Septiembre de 2012
© 1997-2012 Derechos reservados
|
