
|
|||
|---|---|---|---|
|
|
Fisioterapia nas complicações respiratórias da fibrose cística. Relato de caso |
|
|
|
Fisioterapeuta, Centro de Ciências da Saúde e do Esporte - CEFID. Universidade do Estado de Santa Catarina - UDESC. (Brasil) |
Daniela Junckes da Silva Mattos dani_junckes@hotmail.com |
|
|
|
|
|||
|
|
http://www.efdeportes.com/ Revista Digital - Buenos Aires - Año 13 - N° 122 - Julio de 2008 |
|
|
1 / 1
IntroduçãoA fibrose cística (FC) é uma desordem genética autossômica recessiva, causada por mutações no gene que codifica o regulador de condutância transmembrana da FC (BOUCHER, 1998; GOLDMAN e AUSIELLO, 2005; GOSS e BURNS, 2007). Constiui-se em uma anomalia da secreção epitelial do cloro que leva à desidratação das secreções das glândulas exócrinas (POSTIAUX, 2004). É uma doença multissistêmica (JONG et al, 2001), caracterizada por infecções crônicas das vias aéreas, que leva ao desenvolvimento de bronquiectasias, insuficiência pancreática exócrina, disfunções intestinais, anormalidades das glândulas sudoríparas e disfunção genitourinária (BOUCHER, 1998). Para Berhman e Kliegman (2004) o acometimento de diversos órgãos ocorre pela heterogeneidade das mutações e por outros fatores. A incidência estimada é de 1:3.000 nascidos vivos entre caucasianos, caindo para 1:17.000 entre afro-americanos e para 1:90.000 entre asiáticos. Apresenta morbimortalidade muito elevada, com apenas 34% dos pacientes chegando à idade adulta e menos de 10% ultrapassando os 30 anos de idade - a sobrevida média é de 28 anos. (BOUCHER, 1998).
De acordo com Postiaux (2004) desde as primeiras semanas de vida iniciam-se lesões da árvore respiratória e do parênquima pulmonar, sendo a bronquiolite o tipo de lesão pulmonar mais precoce. Geralmente, a manifestação inicial é a tosse, podendo ser acompanhada de sibilos (GOLDMAN e AUSIELLO, 2005). Pode apresentar-se de forma assintomática por longos períodos ou de forma similar a infecções respiratórias agudas prolongadas. Outros podem adquirir uma tosse crônica nas primeiras semanas de vida ou apresentarem pneumonia de repetição. Com a progressão da doença pulmonar, observa-se a não tolerância ao exercício, falta de fôlego e atraso do ganho ponderal ou do crescimento. As exacerbações dos sintomas pulmonares exigem hospitalização para um tratamento eficaz (BEHRMAN e KLIEGMAN, 2004).
Os achados físicos iniciais são o aumento do diâmetro ântero-posterior do tórax, hiper-ressonância generalizada, crepitações grosseiras dispersas ou generalizadas e baqueteamento digital. Pode-se auscultar sibilos expiratórios, especialmente em crianças pequenas. As complicações pulmonares mais comuns incluem atelectasias, hemoptise, pneumotórax e cor pulmonale que aparecem, geralmente, após a primeira década de vida. (BEHRMAN e KLIEGMAN, 2004). Boucher (1998) acrescenta evidência de hiperinsulflação pulmonar. Com mudanças no tórax, pode ser difícil para a criança criar as pressões necessárias e as taxas de fluxo para uma tosse efetiva ou para o aumento da ventilação durante o estresse físico.
Considerando as alterações pulmonares dentre as principais complicações da FC e a importância da atuação fisioterapêutica para controle ou lentificação destas manifestações, o presente estudo tem por objetivo principal descrever o tratamento fisioterapêutico em criança fibrocística durante sua internação hospitalar.
Relato do casoSujeito do sexo feminino, 7 anos, caucasiana, procedente da cidade de Armazém/SC, filha única de pais agricultores, internada na Unidade de Pneumologia para investigação de FC. Hipótese diagnóstica sustentada por déficit de crescimento e nutrição, esteatorréia e má absorção e sintomas respiratórios persistentes. A queixa principal da paciente consistia em dor no peito aos esforços e tosse. A paciente nasceu no ano anterior à inclusão do diagnóstico de FC pelo teste do pezinho, mesmo que sem diagnóstico no período neonatal, as manifestações clínicas iniciaram-se desde o nascimento. Mãe relata diarréias freqüentes com gotículas de gordura, a partir de um ano iniciaram-se infecções em vias áreas superiores (otite, sinusite, bronquite) e tosses diárias com expectoração de coloração amarela, uso freqüente de antibióticos e corticóides. Aos 6 anos consultou pneumo-pediatra para investigação e cinco meses depois foi internada pelo mesmo motivo. O teste para diagnóstico de esteatorréia com corante sudam IV teve resultado positivo, a partir de então, iniciou tratamento prévio medicamentoso e fisioterapêutico (inicialmente 3 vezes por semana, após 2 vezes por semana e atualmente 1 vez por semana e exercícios domiciliares). Em relação à história social, está na fase escolar (2ª série do ensino fundamental), realiza atividades lúdicas compatíveis com a idade, entretanto refere cansaço em prática de brincadeiras que envolvem de médio a grande esforço físico como correr e andar bicicleta. Não apresenta casos semelhantes da doença na família. No leito a paciente estava em bom estado geral, corada e hidratada. Durante avaliação subjetiva, relato de dispnéia com duração de alguns minutos desencadeada por médios e grandes esforços acompanhada de dor torácica, e ainda, tosse produtiva e eficaz freqüente. Ao exame físico identificou-se: baqueteamento digital discreto (Figura 1A), mobilidade torácica presente simétrica bilateralmente, padrão respiratório misto com utilização da musculatura acessória durante a fase inspiratória da respiração, presença de tiragens (subcostal e intercostal) como pode se observar na Figura 1B, ausculta pulmonar com murmúrio vesicular presente e diminuído em ambos hemitórax.
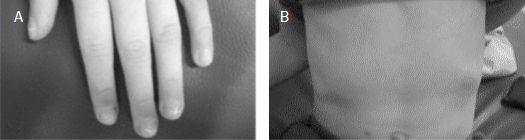
Figura 1. A) Destaque para presença de baqueteamento digital. B) Destaque para tiragem subcostal e intercostal.A avaliação da equipe de nutrição constatou presença de desnutrição aguda. O diagnóstico de fibrose cística foi confirmado após exame de iontoforese do suor com valores de cloro e sódio acima dos valores de referência em três ensaios. Dentre os exames complementares realizou-se: antibiograma pelo qual mostrou ser resistente à penicilina, exame do escarro (bacterioscopia) com poucos leucócitos, teste com Sudam III positivo, esteatócrito igual a 70%, superior do valor de referência (inferior a 4%), leucócitos aumentados (neutrófilos segmentados) e proteínas totais aumentadas (globulina). À radiografia de tórax, hiperinsulflação pulmonar e proeminência hilar com paredes brônquicas espessadas. À ultrassonografia abdominal total, pâncreas mais ecogênico do que o habitual sugerindo FC.
Os objetivos do tratamento consistiram em: aumentar ou manter tolerância ao exercício, melhorar higiene das vias aéreas, aumentar ou manter fluxo aéreo, reexpandir ou manter expansibilidade do pulmão, obter padrão ventilatório normal (diafragmático) e, desta forma, diminuir dor e dispnéia aos médios e grandes esforços ou evitar que sejam desencadeadas em pequenos esforços e prevenir infecções respiratórias. Desta forma, foram realizados: alongamento da musculatura acessória da respiração, manobras de higiene brônquica (MHBs), exercícios respiratórios, exercícios físicos e atividades lúdicas. O tratamento da paciente consistia ainda de: dieta para fibrose cística, nebulização, soro fisiológico e tratamento medicamentoso (dipirona, plasil, oxacilina, ceftazidine, tobramicina, , berotec, atrovent, tobramicina e creon).
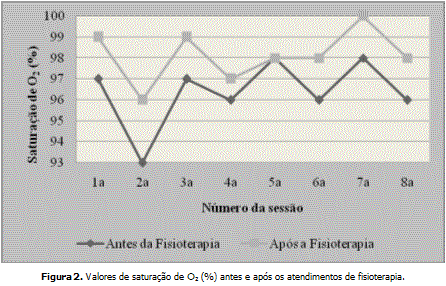
Para prevenção de encurtamentos musculares devido ao uso indevido da musculatura respiratória acessória durante o padrão respiratório misto, foram preconizados alongamentos da musculatura inspiratória da respiração de forma ativo-assistida. Considerando o espessamento do muco no trato respiratório, a presença de tosse diária com expectoração e a prevenção de complicações respiratórias, foram conduzidas MHBs como: vibrocompressão, manobra da aceleração do fluxo expiratório ativo-assistido associado à abertura da glote e técnica de expiração forçada. Paciente apresentou diminuição de secreção das vias aéreas durante as sessões e relato subjetivo de diminuição da tosse. Os exercícios respiratórios foram utilizados com a finalidade de treinar o padrão diafragmático, bem como homogenização do ar no pulmão e reexpansão pulmonar. Outros exercícios respiratórios foram: inspiração e expiração máxima para treino do padrão, inspiração abreviada e desde o volume residual associados à caminhada e propriocepção diafragmática. Para estimular a realização do exercício proposto, foram utilizados inspirômetros de incentivo (Arezee, Voldyne e Respiron). Durante os atendimentos, de forma complementar, realizou-se caminhada, corrida e atividades lúdicas como amarelinha, sempre respeitando os limites do paciente. O tratamento fisioterapêutico em paciente fibrocístico é de longo prazo e de freqüência diária. Programas de tratamento iniciados precocemente têm diminuído complicações da doença. Como o sujeito do estudo obteve diagnóstico aos 7 anos de idade, foram passadas informações em relação à realização do tratamento fisioterapêutico de forma intensiva após o período hospitalar. Como resultados, houve aumento da SapO2 diária (avaliado por oximetria), melhora da higiene brônquica e do padrão respiratório com menos uso da musculatura acessória e aumento da ventilação. Na avaliação subjetiva da paciente obteve-se percepção de melhora da tosse que foram identificados também durante as sessões.
DiscussãoGrande parte da morbidade e mortalidade associada à FC cística está relacionada ao sistema pulmonar, primariamente vias aéreas superiores e inferiores (BELL e ROBINSON, 2007; GOSS e BURNS, 2007). Apesar de o defeito primário da FC estar bem esclarecido em pesquisas científicas, não foram realizados, entretanto, trabalhos completos que aclarem como as mutações originam doenças das vias aéreas na FC. A hipótese mais eminente para explicar este fenômeno é baseada em dados in vitro e propõe que, decorrente da hiperabsorção de sódio e falta de absorção de cloro, o volume do fluido periciliar diminui, com desidratação relativa da camada. Isto leva a um baixo volume de fluido, debilidade na função ciliar e lentificação no transporte mucociliar e, em conseqüência disto, proliferação bacteriana. Durante o processo infeccioso, neutrófilos são recrutados às vias aéreas com subseqüente liberação de citocinas pró-inflamatórias, o que gera um ciclo vicioso de infecção crônica e inflamação e, finalmente, doenças nas vias aéreas (GOSS e BURNS, 2007).
De acordo com Robinon (2001) e Bell e Robinson (2007) houve uma melhora considerável na sobrevivência de fibrocísticos nos últimos 50 anos. Nos anos 40, conseqüências adultas da FC eram mínimas já que a maioria dos pacientes morria durante a infância e início da adolescência. Nos anos 90, por sua vez, a expectativa média de vida em crianças nascidas excedeu os 40 anos, e mais do que 85% das crianças irão alcançar a vida adulta. Sugere-se que diferentes fatores foram responsáveis pela melhora dos resultados clínicos em pacientes com FC, inclui-se: melhora no tratamento de complicações neonatais por íleo-meconial, maior suporte nutricional e formulações mais efetivas de enzimas pancreáticas, advento de novos e mais efetivos agentes mucolíticos, MHBs, tratamentos com antibióticos e o estabelecimento de centros especializados no cuidado de FC. Estas mudanças no tratamento contribuíram para a redução do declínio da função pulmonar tanto em adultos quanto em crianças com FC.
O estado do paciente deve ser regularmente monitorado por meio da avaliação dos sintomas, exame físico e espirometria. A saturação de O2 deve ser mensurada rotineiramente em pacientes com disfunções pulmonares de moderada a severa para avaliar a necessidade de suporte de oxigênio. Uma avaliação microbiológica completa do escarro e teste de sensibilidade ao antibiótico devem ser realizados no mínimo uma vez ao ano (YANKASKAS et al., 2004).
Em relação ao tratamento de fibrocísticos as medidas-chave são antibioticoterapia, higiene brônquica e suporte nutricional em adultos e crianças (YANKASKAS et al., 2004). Conforme destaca Holland et al. (2003), o tratamento fisioterapêutico deve ser integral para cuidado das alterações pulmonares da FC. Mostrou-se que manobras para auxiliar a remoção de secreções das vias aéreas diminuíram a velocidade de deterioração da função pulmonar e há necessidade destas manobras tanto em períodos de exacerbação quanto de estabilidade. Winden et al. (1998) afirmam que a expectoração do escarro, auxiliada pela fisioterapia, por meio de drenagem postural e tosse - denominadas fisioterapia convencional - foram terapias padrões durante anos. Ciesla (1996) observou efeitos benéficos na oxigenação arterial após realização de manobras desobstrutivas em pacientes internados em Unidade de Terapia Intensiva (UTI). A utilização de incentivadores respiratórios em crianças é preconizada como técnica auxiliar na reexpansão pulmonar, pois fornece ao paciente o feedback da sua realização através do número de esferas elevadas a cada inspiração e é amplamente usada no ambiente hospitalar como forma profilática das complicações pulmonares. Com o passar dos tempos, os pacientes tornam-se mais independentes e freqüentemente procuram técnicas de higiene que possam ser realizadas sem assistência. Técnicas fisioterapêuticas auto-administradas incluem: técnica de expiração forçada, ciclo ativo da expiração, drenagem autogênica e o uso de máscara de pressão positiva expiratória (WINDEN et al., 1998; YANKASKAS et al., 2004).
Ensaios clínicos adicionais são necessários para estabelecer um programa ótimo de higiene das vias aéreas. A expectoração de catarro foi o primeiro resultado observado na maioria dos ensaios, entretanto é um marcador não confiável da eficácia. A preservação da função pulmonar é uma evidência mais convincente da eficácia (YANKASKAS et al., 2004).
No que diz respeito à atividade física, deve ser vista como uma importante técnica complementar às descritas anteriormente já que aumenta higiene das vias aéreas. Um ensaio clínico randomizado demonstrou que exercícios aeróbicos regulares atenuam o declínio da função pulmonar em mais do que 3 anos quando comparado com grupo controle. Adicionalmente, exercício físico vigoroso aumenta atividade cardiovascular, capacidade funcional e qualidade de vida. O nível de atividade física medida pelo O2 exalado é correlacionado com a sobrevida em FC. Por essas razões, com exceção àqueles com impedimento pela condição clínica, todos os adultos com FC devem ser encorajados a exercitar-se. Idealmente, os pacientes devem aprender técnicas de exercício sob supervisão de fisioterapeuta qualificado. Atividades aeróbicas como natação, corrida e ciclismo são as formas mais recomendadas de exercício (YANKASKAS et al., 2004).
A equipe de saúde deve assistir o paciente e a família em programas de higiene que melhor se adequem ao estilo de vida e atividades do paciente. Tipicamente, consiste em técnicas convencionais e/ou alternativas de higiene brônquica combinada com exercícios aeróbicos. A freqüência e duração de cada tratamento deve ser individualizada. Pacientes com sintomas de mínimo a médios necessitam de sessões diárias, enquanto aqueles com maior volume de secreção necessitam três ou mais sessões pó dia. (YANKASKAS et al., 2004).
Em conjunto com a atuação de outros profissionais da equipe de saúde e a terapia medicamentosa, a participação da Fisioterapia exerce papel fundamental para a lentificação das complicações pulmonares. Desta forma, contribui para um melhor prognóstico da doença.
Referências bibliográficas
AZEREDO, C. A. Fisioterapia Respiratória Moderna. 4 ed. São Paulo: Manole, 2002. p. 44-49.
BELL, S. C.; ROBINSON, P. J. Exacerbations in cystic fibrosis: 2 Prevention. Thorax. v. 62, p. 723-732, 2007.
BEHRMAN, R.; KLIEGMAN, R. Nelson: Princípios de pediatria. 4 ed. Rio de Janeiro: Guanabara Koogan, 2004. 932 p.
BOUCHER, R. C. Cystic Fibrosis. In Fauci A. S., et al. Harrison's Principles of Internal Medicine. 14 ed. 1998.
CIESLA, N. D. Chest Physical therapy for patients in the Intensive Care Unit. Physical Therapy. v. 76, p. 609-625, 1996.
GOLDMAN, L.; AUSIELLO, D. Tratado de medicina interna. São Paulo: Elsevier, 2005. 3000 p.
GOSS, C. H; BURNS, J. L. Exacerbations in cystic fibrosis: Epidemiology and pathogenesis. Thorax. v. 62, p. 360-367, 2007.
HOLLAND, A. E.; DENEHY, L.; NTOUMENOPOULOS, G.; NAUGHTON, M. T.; WILSON, J. W. Non-invasive ventilation assists chest physiotherapy in adults with acute exacerbations of cystic fibrosis. Thorax. v. 58, p. 880-884, 2003.
JONG, W.; VAN AALDEREN, W. M. C.; KRAAN, J.; KOËTER, G. H.; VAN DER SCHANS, C. P. Inspiratory muscle training in patients with cystic fibrosis. Respiratory medicine. v. 95, p. 31-36, 2001.
POSTIAUX, G. Fisioterapia respiratória pediátrica: o tratamento guiado por ausculta pulmonar. 2 ed. Porto Alegre: Artmed, 2004. 301 p.
ROBINSON, P. Paediatric origins of adult lung disease. Thorax. v. 56, p. 237-241, 2001.
TECKLIN, J. S. Fisioterapia pediátrica. São Paulo: Artmed, 2001. 480 p.
WINDEN, C. M. Q.; VISSER, A.; HOP, W.; STERK, P. J.; BECKERS, S.; JONGSTE, J. C. Effects of flutter and PEP mask physiotherapy on symptoms and lung function in children with cystic fibrosis. Eur Respir J. v. 12, p. 143-147, 1998.
YANKASKAS, R.; MARSHALL, B. C.; SUFIAN, B.; SIMON, R. H.; DAVID, R. Cystic fibrosis adult care: consensus conference report. Chest. v. 125, p. 1S-39S, 2004.
 |
|
|---|---|
|
revista
digital · Año 13
· N° 122 | Buenos Aires,
Julio 2008 |
|